advertisement


Glaucoma Overview
Pigment Dispersion Syndrome

Tanuj Dada
The association of ocular pigment dispersion with elevated intraocular pressure (IOP) was first described by Von Hippel1 in the early years of the nineteenth century and the clinical entity was later termed as pigment dispersion syndrome (PDS) and pigmentary glaucoma (PG) by Sugar.2 PDS is characterized by a triad of increased trabecular meshwork (TM) pigmentation, iris transillumination defects and pigment granules on the corneal endothelium (Krukenberg spindle). It affects nearly 2-3%3 of the Caucasian population undergoing glaucoma screening and the conversion rate from PDS to PG has been estimated to be 10% after five years and 15% after 15 years.4 The disease is more common in males, is associated with mild-moderate myopia and nearly one-third of PDS patients eventually develop high ocular hypertension (IOP) or optic nerve damage (glaucoma).
Pathophysiology
PDS eyes are characterized by a deep anterior chamber and a concave iris configuration associated with posteriorly inserted iris, floppy iris stroma or abnormal posterior pigmented epithelium of iris, long anterior zonules, and a relatively mobile lens.5 On blinking, aqueous is pushed from the posterior to the anterior chamber, thus creating a pressure gradient.6 During exercise and accommodation,7,8 the lens moves forward increasing the area of irido-lenticular contact which acts as a flap valve allowing movement of aqueous only in one direction ‒ from posterior to anterior chamber, raising the aqueous pressure in the anterior chamber, causing posterior bowing of the iris and a resulting in ‘reverse pupillary block’. This further aggravates the iridozonular friction and promotes release of iris pigments into the aqueous. A developmental or a congenital anomaly9 in pigmented epithelial cells of iris and ciliary body making them more vulnerable to the irido-zonular rubbing has also been advocated as another possible mechanism for pigment dispersion.
The iris pigments thus released, gets deposited in the anterior segment of the eye including the angle structures, and clogs the trabecular meshwork leading to a decrease in the facility of outflow. The uveoscleral outflow is not affected in PDS/PG, unlike in ocular hypertension (OHT)/primary open-angle glaucoma (POAG). In the chronic stages, the TM endothelial cells are overburdened with phagocytosis of melanin granules, eventually leading to cell death. TM beams are then left bare causing collapse and fusion of the TM, increased outflow resistance and chronically raised IOP with consequent optic neuropathy (Fig. 1).9
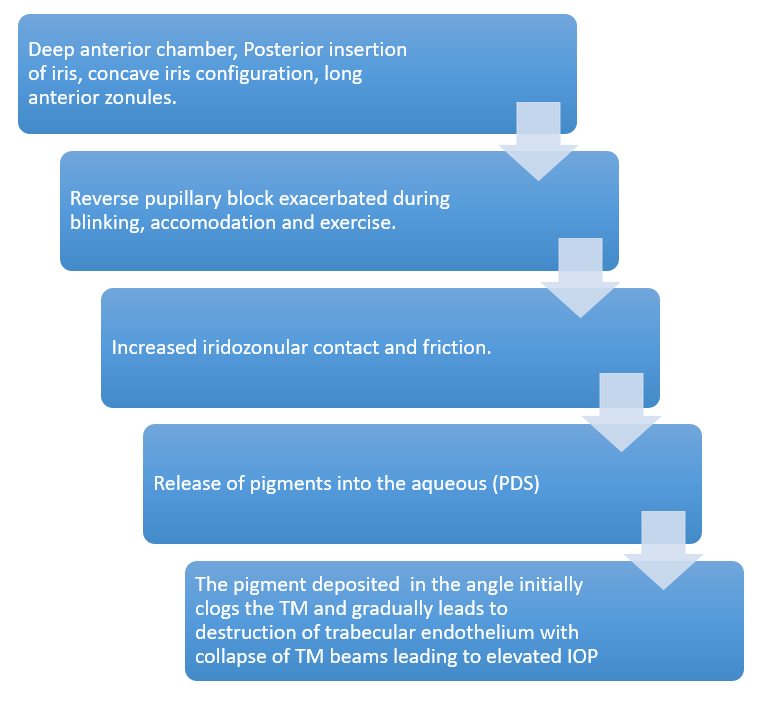
Fig 1: Flowchart summarizing Pathophysiology of PDS
and PG.
Clinical features
PDS is a bilateral disease with age of onset of pigmentary glaucoma usually being in the third to fifth decade of life, while signs of PDS may be visible many years before glaucoma sets in.
Symptoms
Patients with PG may present with a history of colored halos and episodic blurring of vision due to an acute rise in IOP, commonly reported after prolonged accommodative effort, working in dim light or after physical exercise. Many patients may present only in an advanced stage with symptoms similar to POAG. It may be detected an incidental finding during the routine ophthalmic examination, often for associated myopia.
Signs
The pigments released are dispersed by aqueous currents and deposited throughout the anterior segment, giving rise to characteristic clinical features:
- Krukenberg spindle: Vertical spindle-shaped deposition of iris pigments on corneal endothelium, infero-central in location corresponding to the convection currents of aqueous. (Fig. 2a)
- Increased TM pigmentation: A homogeneous dark brown band of pigmentation, especially in the superior angle with concave iris configuration is visible on gonioscopy. The Schwalbe’s line has pigmented deposition and is known as Sampaolesi’s line. (Fig. 2b, 2c)
- Iris transillumination defects: When the iris is examined in retro-illumination, radial spoke-like transillumination defects are visible in the mid-peripheral iris. (Fig. 2d)
- Pigment deposition on anterior iris surface: Pigments get deposited in the iris furrows as concentric rings. This feature again is not easily appreciated in dark-colored irides. In cases with asymmetric presentation, iris heterochromia and anisocoria (due to dilator atrophy) may be evident.
- Free-floating pigment granules in the aqueous.
- Pigment deposition on the anterior and posterior lens capsule.
- Zentamayer ring or Scheie strip: Pigments may be deposited in a ring-like fashion at the level of insertion of zonules into the posterior lens capsule. This is best visualized after pupillary dilation with the patient looking in upgaze.
- Egger’s line: Refers to the deposition of pigments on the anterior hyaloidocapsular ligament.
- Posterior segment: Patients with PDS have an increased frequency of peripheral retinal degenerations like lattice degeneration and retinal breaks. Rhegmatogenous retinal detachment is seen in 6-7% cases of PDS, irrespective of the amount of myopia. A dilated peripheral retinal screening is essential.
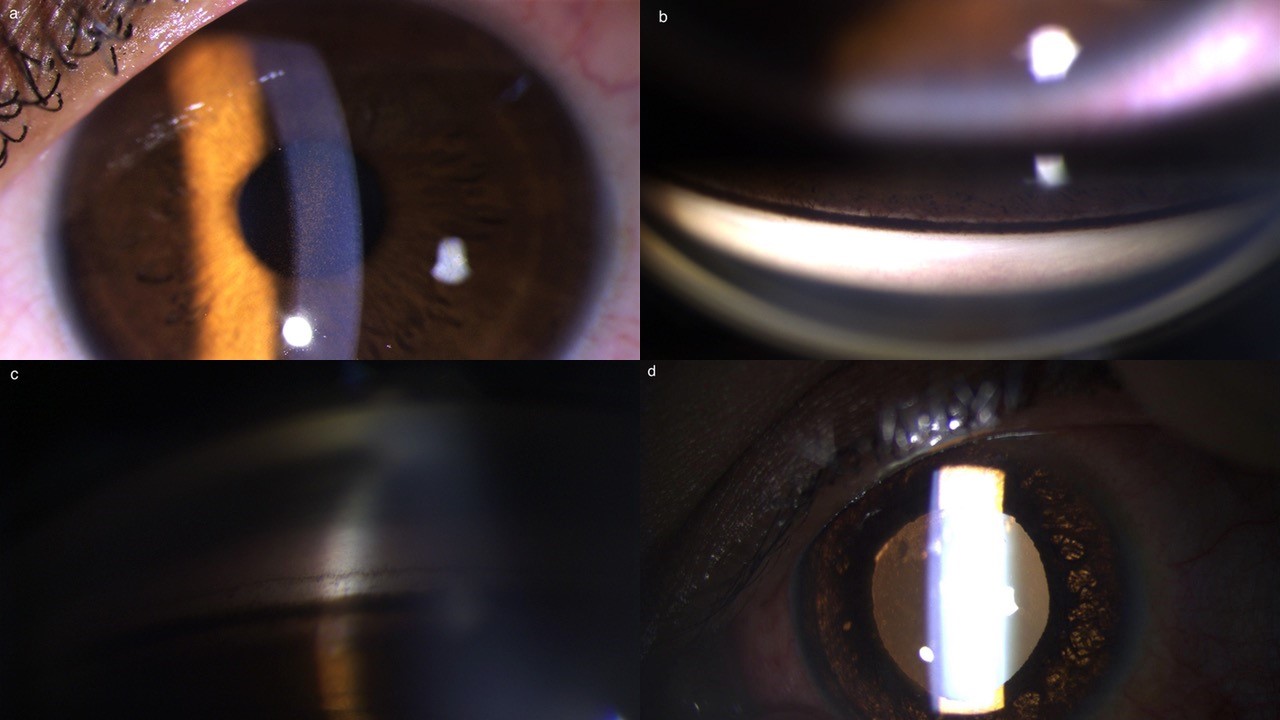
Fig 2: Clinical features of Pigment dispersion syndrome.
(a) Krukenberg spindle (b) Gonioscopy shows dense trabecular meshwork pigmentation
in superior angle. (c) Concave iris with angle pigmentation & Sampaolesi line.
(d) Mid-peripheral iris defects in iris on retro-illumination.
The disease has four distinct clinical phases (Fig. 3). The final ‘regression phase’ is a less-commonly diagnosed entity. In some patients, the pigment begins to clear up from the anterior segment and IOP may return to normal. However, the glaucomatous optic nerve head changes remain, leading to a misdiagnosis of normal-pressure glaucoma. The increased pigmentation of superior vs. inferior angles visible on gonioscopy, may provide a vital clue to diagnose a regressed PDS.
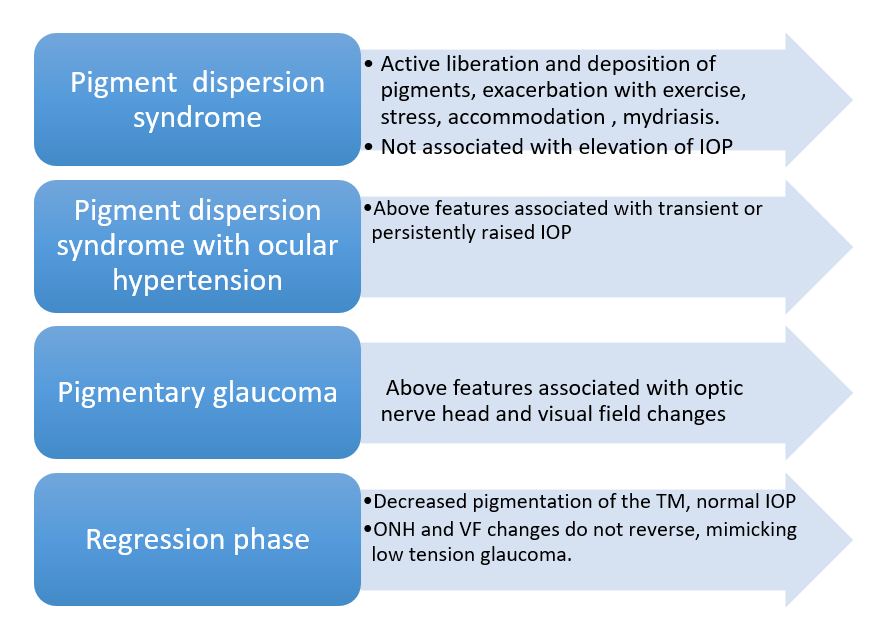
Fig 3: Clinical phases of the disease.
Diagnosis
Clinical
PDS and PG can be easily diagnosed with careful clinical examination to look for Krukenberg spindle, iris transillumination defects and pigments on lens capsule. Gonioscopy may show dense pigment deposition and concave iris configuration, thus providing a clinching diagnosis.
Provocative test
Mydriasis with Phenylephrine leading to release of ten or more pigment particles with/without an acute rise in IOP indicates an active phase of pigment dispersion. As the dilator pupillae contracts, the pigmented cells of posterior iris surface rupture releasing large amounts of pigment in the aqueous. This pigment clogs the TM and causes a transient and acute rise in IOP. Although this test can be used to identify patients with active disease and ‘high risk’ for progression, this test does not have an absolute positive predictive value and may ‘miss’ high-risk patients.
Imaging
Ultrasound biomicroscopy (UBM) and anterior segment OCT (Fig. 4) may aid the diagnosis of PDS. Characteristic posterior bowing of the iris, amount of irido-lenticular and irido-zonular contact can be seen. It can also be used to study the changes in iris configuration before and after laser iridotomy.
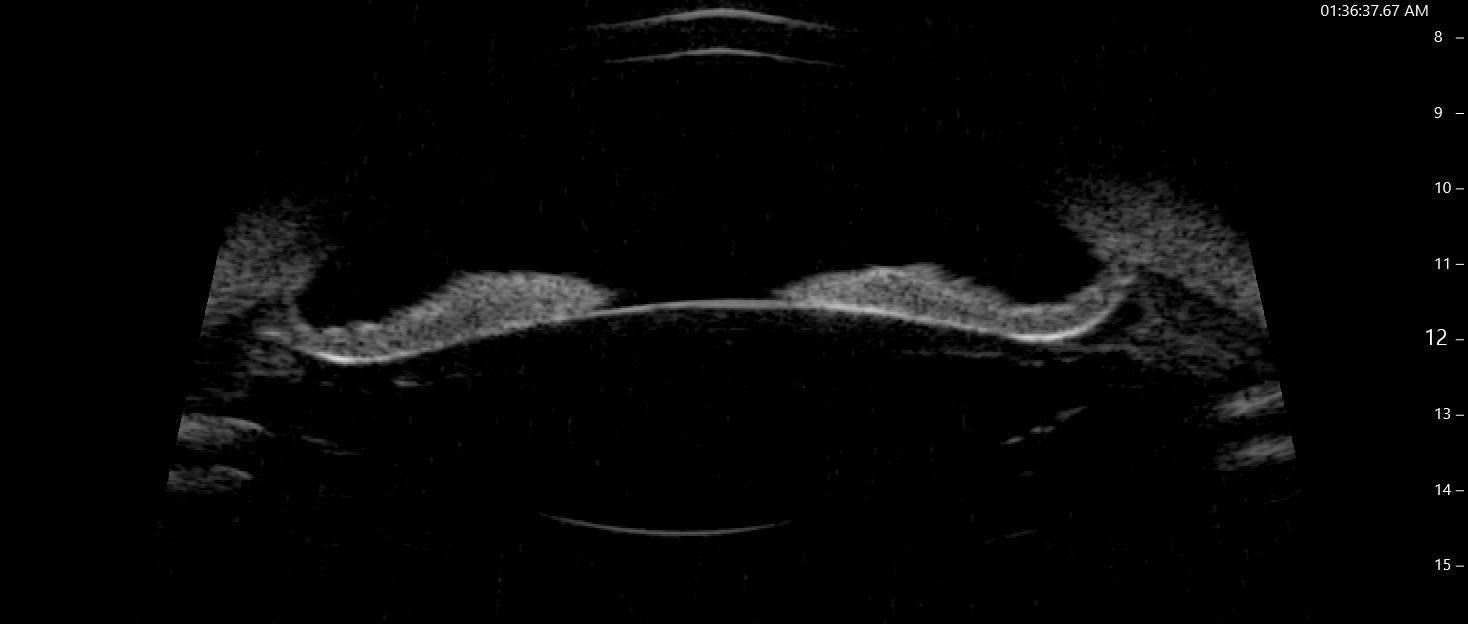
Fig.4: UBM image showing posterior bowing of mid-peripheral
iris.
Differential diagnosis
All conditions that cause liberation of pigments and deposition in various structures of the anterior segment form a differential diagnosis for PDS. Some of these include anterior uveitis (look for cells vs. pigment, keratic precipitates, posterior synechiae), pseudoexfoliation (look for pseudoexfoliative material and peri-pupillary transillumination defects), trauma, malpositioned IOL and neoplasm of the iris/ciliary body (melanoma).
Management
The management protocol of pigmentary glaucoma is essentially same as POAG. Although pilocarpine causes miosis and can reduce irido-zonular contact and increases TM outflow, it is poorly tolerated in the young myopic population of PDS and increases risk of retinal detachment. Prostaglandin analogues which work by enhancing outflow, are the preferred first line agents for lowering of IOP. Other classes of drugs like beta blockers, alpha agonists and carbonic anhydrase inhibitors can be added as second-line therapy, although decreasing aqueous production and inflow may promote pigment accumulation.
Laser peripheral iridotomy
Patients who test positive on phenylephrine mydriatic test are most likely to benefit with an laser peripheral iridotomy (LPI).10 An iridotomy only relieves the reverse pupillary block, though some patients will continue to have concave iris configuration and IOP spikes after prolonged accommodation or exercise. Even after LPI, the risk of developing PG is not completely mitigated and requires regular long-term follow-up. In a ten-year follow-up study, Gandolfi et al. reported that when left untreated, more than 60% of high-risk PDS eyes (positive phenylephrine test) developed ocular hypertension as compared to only 10% of low-risk PDS eyes (negative phenylephrine test). In the high-risk PDS eyes, only 14.3% of LPI treated eyes developed ocular hypertension (> 5 mmHg) as compared to 61.9% of untreated eyes. Hence LPI significantly lowered the risk of development of ocular hypertension in PDS patients demonstrating active pigment dispersion. However, LPI is only effective in early phase of the disease and once ocular hypertension/glaucoma has already set in, it will have a limited role. LPI in PDS can be associated with a significant release of pigment and high IOP spikes, with potential of worsening the disease itself. A technique using trans-illumination with reduced requirement of laser energy can be an option to reduce this complication.11
Laser trabeculoplasty
Open angles with increased pigmentation makes these patients suitable candidates for laser trabeculoplasty. Caution must be taken to use low levels of energy, and preoperative use of alpha agonists to prevent IOP spikes.
Surgery
If target IOP is not achieved with medical therapy and/or laser trabeculoplasty, filtration surgery is the next course of intervention. Use of adjunctive antifibrotic agents is recommended as the patients undergoing surgery are young, although the risk of hypotonic maculopathy in myopes warrants a cautious use of these agents. There should be a low threshold for cataract surgery if patients present with lenticular opacification, as a lens extraction along with irrigation aspiration may help to decrease the quantum of pigment accumulated in the angle and additionally remove irido-lenticular contact, preventing further disease progression.
Summary
Pigment dispersion can be diagnosed with a careful anterior segment slit lamp biomiscroscopic evaluation including gonioscopy. An early diagnosis of PDS and reduction of active pigment release due to iridozonular friction by performing a laser peripheral iridotomy is recommended. Once PDS is associated with ocular hypertension or optic neuropathy, reduction of IOP using laser trabeculoplasty and prostaglandin analogues is appropriate. An examination of the retinal periphery for lesions predisposing to retinal detachment must be performed in all PDS patients; other causes of pigment dispersion must be ruled out (especially in case of asymmetric/unilateral disease) and long-term follow-up is required, until disease remission is documented. The research question for the future is the role of lens extraction with or without MIGS in prevention of disease progression in PDS and treatment of pigmentary glaucoma.
References
- Von Hippel E. Zur pathologischen Anatomie des Glaukom. Arch Ophthalmol 1901;52:498.
- Sugar HS. Pigmentary glaucoma. A 25-year review. Am J Ophthalmol 1966;62(3):499-507.
- Ritch R, Steinberger D, Liebmann JM. Prevalence of pigment dispersion syndrome in a population undergoing glaucoma screening. Am J Ophthalmol 1993;115(6):707-710.
- Siddiqui Y, Ten Hulzen RD, Cameron JD, et al. What is the risk of developing pigmentary glaucoma from pigment dispersion syndrome? Am J Ophthalmol 2003;135:794-799.
- Potash SD, Tello C, Liebmann J, Ritch R. Ultrasound biomicroscopy in pigment dispersion syndrome. Ophthalmology 1994;101(2):332-339.
- Liebmann JM, Tello C, Chew SJ, Cohen H, Ritch R. Prevention of blinking alters iris configuration in pigment dispersion syndrome and in normal eyes. Ophthalmology 1995;102(3):446-455.
- Pavlin CJ, Macken P, Trope GE, Harasiewicz K, Foster FS. Accommodation and iridotomy in the pigment dispersion syndrome. Ophthalmic Surg Lasers 1996;27(2):113-120.
- Schenker HI, Luntz MH, Kels B, Podos SM. Exercise-induced increase of intraocular pressure in the pigmentary dispersion syndrome. Am J Ophthalmol 1980;89(4):598-600.
- Gottanka J, Johnson DH, Grehn F, Lütjen-Drecoll E. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. J Glaucoma 2006;15(2):142-151.
- Gandolfi SA, Ungaro N, Tardini MG, et al. A 10 year follow up to determine the effect of YAG laser iridotomy on the natural history of PDS: a RCT. JAMA Ophthalmol 2014;132(12):1433-1438.
- Karickhoff JR. Pigmentary dispersion syndrome and pigmentary glaucoma: a new mechanism concept, a new treatment, and a new technique. Ophthalmic Surg 1992;23(4):269-277.
